Laboratory of Molecular and Cellular Biology of Cancer
Hôpital Kirchberg9, Rue Edward Steichen
L-2540 Luxembourg - Luxembourg
Site web - marc.diederich@lbmcc.lu -
Principal investigator
Pr Marc Diederich PhD, directeur du LBMCC

Research themes
The Laboratory of Molecular and Cellular Cancer Biology (LBMCC) investigates molecular mechanisms involved in cancer, inflammation, and immunogenicity, in solid and hematological cancer. Among multiple mechanistic approaches, we integrate the investigation of compound-mediated cell death and differentiation mechanisms with autophagy and its modulation into our projects (1-4):
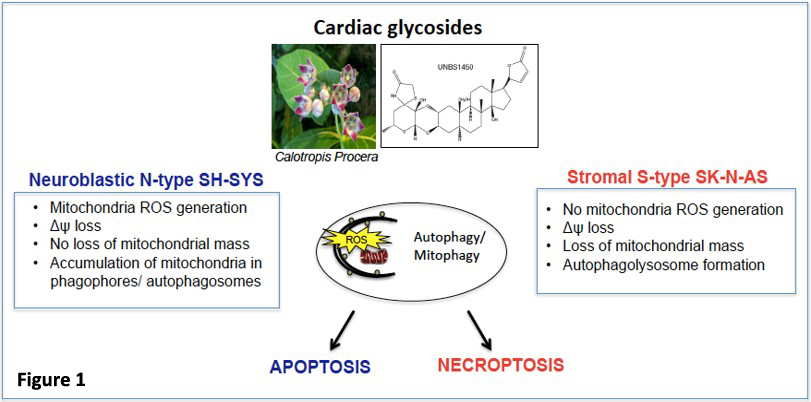
We identify biomarkers and pathways controlling the mitochondrial turnover by mitophagy or the autophagic flux in cancer cells. We investigate the potential anti-cancer strategies centered on targeting deregulated mitochondria in neuroblastoma tumors, to define biomarkers predicting treatment response. We aim to identify novel, more effective therapies and combinational personalized treatment approaches for high-risk neuroblastoma sub-groups. A previous study identified mitophagy as a determinant for the anti-cancer activity of cardiac glycosides in different neuroblastoma subtypes. We demonstrated that the hemi-synthetic cardiac glycoside UNBS1450 exerts its anti-cancer activity primarily through the early induction of mitophagy in response to a severe mitochondrial stress response, in turn promoting cell type-specific demise (Figure 1) (5).
On the other hand, we aim to predict cell markers and pathways controlling the autophagic flux as well as pharmacological compounds acting as perturbators of autophagy. We study the cell death-sensitizing effects of autophagy after targeting various cancer cell models with single or combinational regimens including newly synthesized or purified compounds. Recently we investigated a novel class of microtubule-altering agents derived from garlic polysulfanes in colon cancer with differential genetic backgrounds (6). We also examined the inhibition of autophagic flux by cardiac glycosides (7-9). Recently, in lung cancer, we showed that autophagy modulation correlated with the genetic background of cancer subtypes hinting at improved patient stratification (10).
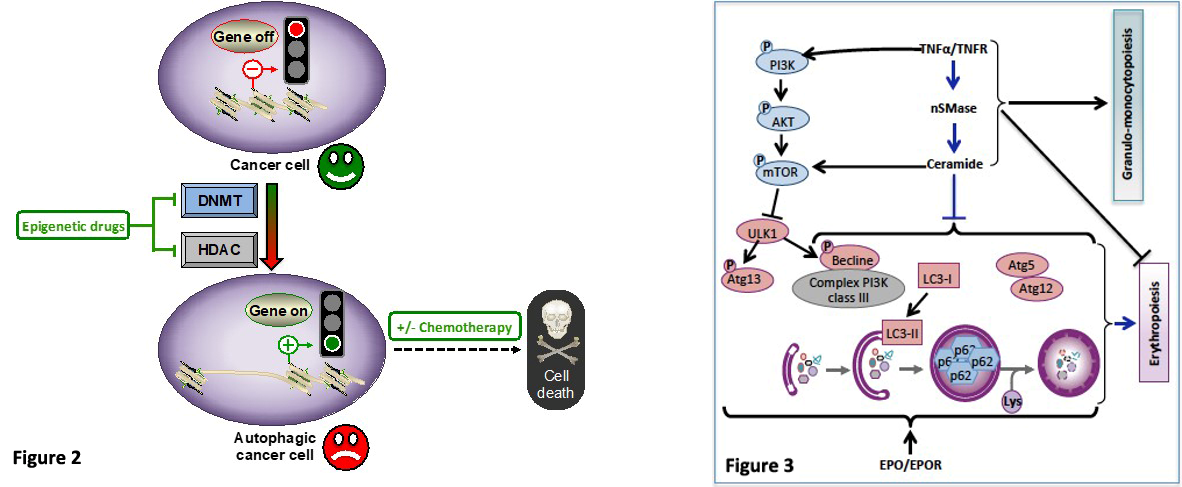
In line with the pharmacological modulation of autophagy, we also identified epigenetic targeting of autophagy as an effective anticancer strategy. Epigenetic mechanisms represent a significant component of the autophagic process and therefore have the potential to influence autophagy-related cellular decisions of life and death. Pathological epigenetic modifications play a critical role in tumorigenesis associated with a deregulation of networks (3) and mechanisms controlling autophagy. Hence, we aim to reprogram cancer cells with epigenetic drugs such as DNA methyltransferase (DNMT) and/or histone deacetylase (HDAC) inhibitors alone or in combination with other chemotherapeutic agents to impair or exacerbate the autophagy flux, with the goal to improve anti-cancer activities (Figure 2). For instance, we showed that prolonged exposure of chronic myeloid leukemia (CML) cells to the clinically approved hypomethylating agent 5-aza-2’-deoxycytidine induces autophagy and leads subsequently to mitochondrial-dependent apoptosis. Such a therapeutic approach sensitizes CML cells to synergistic apoptosis induction in combination with either conventional cell death inducers or approved HDAC inhibitors (11). More recently, we identified isofistularin-3, a new DNMT inhibitor extracted from a marine sponge, which triggers autophagy in lymphoma cells and sensitizes them to TRAIL, an apoptotic inducer (12, 13). Altogether, our findings point out the interest to modulate autophagy in cancer cells as a potent therapeutic approach. We further aim to characterize the underlying mechanisms and molecular determinants of such treatments with the ultimate goal of delivering improved therapeutic choices for cancer patients (14).
Beyond the pharmacological modulation of autophagy, we also characterize the cellular and molecular mechanisms involved in chronic inflammation-mediated inhibition of erythropoiesis leading anemia in cancer patients. The pro-inflammatory cytokine TNFα inhibits erythropoiesis and causes anemia in patients with cancer and chronic inflammatory diseases (15). Autophagy plays an essential role during erythropoiesis (16). We studied the impact of the TNFα/ceramide pathway on autophagy in erythropoietin (Epo)-stimulated CD34+ hematopoietic stem cells (HSC). We demonstrate that the TNFα/ceramide pathway inhibits erythropoiesis and concomitantly induces myelopoiesis via inhibition of late steps of autophagy. Indeed, the TNFα/ceramide pathway inhibited autophagy in Epo-induced CD34/HSCs by inducing phosphorylation of mTORS2448 and ULK1S758, thus inhibiting Atg13S355 phosphorylation. Autophagosome formation was blocked as shown by transmission electron microscopy and GFP-LC3 punctae formation. Furthermore, the TNFα/ceramide pathway inhibited the expression of beclin 1 and Atg5-Atg12 complex formation. Rapamycin prevented the inhibitory effect of TNFα and ceramides on erythropoiesis while inhibiting induction of myelopoiesis. In contrast, bafilomycin A1, but not an anti-Atg5 siRNA, induced myeloid differentiation, while both impaired erythropoiesis. Altogether, our results revealed an essential role of autophagy in erythroid vs. myeloid differentiation (Figure 3) (17).
Descriptive figure
Publications
(1) Cerella C., et al., Biotechnol Adv, 2014, 10.1016/j.biotechadv.2014.03.006
(2) Diederich M. and Cerella C., Semin Cancer Biol, 2016, 10.1016/j.semcancer.2016.06.001
(3) Morceau F., et al., Biotechnol Adv, 2015, 10.1016/j.biotechadv.2015.03.013
(4) Schnekenburger M., et al., Biotechnol Adv, 2014, 10.1016/j.biotechadv.2014.03.009
(5) Radogna F., et al., Oncogene, 2016, 10.1038/onc.2015.455
(6) Yagdi Efe E., et al., Cancer Lett, 2017, 10.1016/j.canlet.2017.09.011
(7) Diederich M., et al., Biochem Pharmacol, 2017, 10.1016/j.bcp.2016.08.017
(8) Juncker T., et al., Biochem Pharmacol, 2011, 10.1016/j.bcp.2010.08.025
(9) Cerella C., et al., Cell Death Dis, 2015, 10.1038/cddis.2015.134
(10) Schneider N. F. Z., et al., Front Pharmacol, 2018, 10.3389/fphar.2018.00070
(11) Schnekenburger M., et al., Biochem Pharmacol, 2011, 10.1016/j.bcp.2010.10.013
(12) Florean C., et al., Mar Drugs, 2018, 10.3390/md16120518
(13) Florean C., et al., Oncotarget, 2016, 10.18632/oncotarget.8210
(14) Radogna F., et al., Biochem Pharmacol, 2015, 10.1016/j.bcp.2014.12.018
(15) Chateauvieux S., et al., Biochem Pharmacol, 2011, 10.1016/j.bcp.2011.06.045
(16) Orsini M., et al., Biochem Pharmacol, 2018, 10.1016/j.bcp.2018.04.007
(17) Orsini M., et al., Cell Death Differ, 2018, 10.1038/s41418-018-0245-x
Composition de l'équipe
Dr Franck Morceau, PhD, chef d’équipe, franck.morceau[at]lbmcc.lu
Dr Claudia Cerella, PhD, chef d’équipe, claudia.cerella[at]lbmcc.lu
Dr Michael Schnekenburger, PhD, chef d’équipe, michael.schnekenburger[at]lbmcc.lu
Dr Flavia Radogna, PhD, postdoc, flavia.radogna[at]lbmcc.lu
Dr Cristina Florean, postdoc, cristina.florean[at]lbmcc.lu
Dr Esma Yagdi, PhD, ancienne doctorante
Dr Marion Orsini, PhD, ancienne doctorante