Signaling and pathophysiology of heart failure and aging
CHU Rangueil, 1 avenue du Pr Jean Poulhès - ToulouseSite web - jeanne.perez@inserm.fr -
Principal investigator
Frank Lezoualc’h et Jeanne Mialet-Perez

Research themes
More than one million of french people suffer from heart failure (HF), a condition for which there is currently no therapy. HF is characterized by oxidative stress, energetic deficiency, cardiomyocyte hypertrophy/death, and development of interstitial fibrosis. Mitochondrial dysfunction plays a major role in the development of HF and recent studies have highlighted the importance of effective “mitochondrial quality control” to limit oxidative damage during cardiac stress [1]. Restoration of the autophagy-lysosome pathway, and more particularly mitophagy, is thus emerging as a new therapeutic axis in HF in order to counter oxidative stress, energetic deficiency and cardiomyocyte death [2]. At present, many contradictions remain in the understanding of the mechanisms underlying the alterations of autophagy during cardiac stress. In our team, we are studying the molecular mechanisms leading to cardiac pathologies and aging, in order to identify new therapeutic targets and to evaluate new pharmacological interventions to limit the development of HF (Figure 1). One of our objectives is to better understand how these molecular events interfere with autophagy to regulate the fate of cardiomyocytes (cell death, hypertrophy, senescence). By improving autophagy, we aim at counteracting the deleterious mechanisms associated with cardiac dysfunction including cell death and mitochondrial damage.
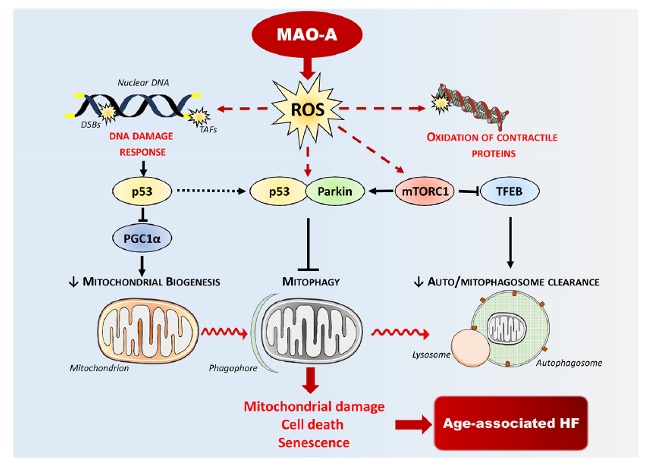
Among these mechanisms, we have shown that the cAMP-sensitive protein Epac1 promotes autophagy at the early stages of cardiac remodeling [3]. In addition, we have characterized a new signaling pathway coupling the production of reactive oxygen species (ROS) by the enzyme monoamine oxidase-A (MAO-A) to mitophagy (Figure 2). MAOs are mitochondrial enzymes that produce H2O2 upon degradation of their substrates, generating mitochondrial oxidative stress and cardiomyocyte death/senescence [4, 5]. Chronic activation of cardiac MAO-A during aging inhibits mitophagy via the protein parkin and amplifies the mitochondrial damage observed in this context [6, 7]. Part of our work is also aimed at better understanding the close link between the accumulation of autophagic vesicles and mitochondrial dysfunction in HF. Importantly, we have established that MAO-A overactivation leads to an alteration of lysosomal acidification associated with a defect in the nuclear translocation of the transcription factor TFEB, a “master regulator” of autophagy and lysosome biogenesis (Figure 2). In vitro, we have shown that the direct consequences of these alterations were a blockage of the autophagic flux, participating in cardiomyocyte necrosis [8]. Very interestingly, overexpression of TFEB in mice reduced the accumulation of autophagosomes, the mitochondrial fission and cardiomyocyte death and prevented the development of HF in these mice. Thus, a new line of research is moving towards restoring “autophagic flux” in HF, via an improvement in autophagosome-lysosome fusion and/or lysosomal function. Some polyphenols seem interesting in this area, via their activating role of TFEB, and some of our work has shown that Oleuropein aglycone, a polyphenol extracted from extra virgin olive oil, is a potential candidate [9]. On the other hand, there is still a drastic lack of lysosomotropic tools able to restore lysosome function in a pathological context, and we are now actively working on this topic.
Descriptive figure

Publications
1. Brown, D.A., et al., Mitochondrial function as a therapeutic target in heart failure. Nature Reviews Cardiology, 2017. 14(4): p. 238-250.
2. Mialet-Perez, J. and C. Vindis, Autophagy in health and disease: focus on the cardiovascular system. Essays in biochemistry, 2017. 61(6): p. 721-732.
3. Laurent, A.-C., et al., Exchange protein directly activated by cAMP 1 promotes autophagy during cardiomyocyte hypertrophy. Cardiovascular research, 2015. 105(1): p. 55-64.
4. Santin, Y., et al., Monoamine oxidases in age-associated diseases: New perspectives for old enzymes. Ageing Res Rev, 2021. 66: p. 101256.
5. Santin, Y., et al., Mitochondrial 4-HNE derived from MAO-A promotes mitoCa(2+) overload in chronic postischemic cardiac remodeling. Cell Death Differ, 2020. 27(6): p. 1907-1923.
6. Anderson, R., et al., Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. Embo j, 2019. 38(5).
7. Manzella, N., et al., Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging cell, 2018. 17(5): p. e12811.
8. Santin, Y., et al., Oxidative Stress by Monoamine Oxidase-A Impairs Transcription Factor EB Activation and Autophagosome Clearance, Leading to Cardiomyocyte Necrosis and Heart Failure. Antioxid Redox Signal, 2016. 25(1): p. 10-27.
9. Miceli, C., et al., Oleuropein Aglycone Protects against MAO-A-Induced Autophagy Impairment and Cardiomyocyte Death through Activation of TFEB. Oxidative medicine and cellular longevity, 2018. 2018: p. 8067592.
Composition de l'équipe
Jeanne Mialet-Perez, CRCN INSERM,
Frank Lezoualc’h, DR INSERMCaroline Conte, MCU
Yannis Sainte-Marie, MCU
Olivier Lairez, PU-PH
Yohan Santin, Post-doc
Karina Formoso, Post-doc
Maximin Détrait, Post-doc
Dorian Bergonnier, Ass. Ing.
Jessica Resta, Doctorante
Loubna Kehal, Doctorante