Signalisation et physiopathologie de l’insuffisance cardiaque et du vieillissement
CHU Rangueil, 1 avenue du Pr Jean Poulhès - ToulouseSite web - jeanne.perez@inserm.fr -
Responsable d'équipe
Frank Lezoualc’h et Jeanne Mialet-Perez

Thématiques de recherche
Plus d’un million de Français souffrent d’insuffisance cardiaque (IC), une pathologie dont il n’existe à l’heure actuelle aucun traitement curatif. L’IC se caractérise par un stress oxydant, une déficience énergétique, une hypertrophie/mort des cardiomyocytes et l’installation de fibrose interstitielle. La dysfonction mitochondriale joue un rôle prépondérant dans le développement de l’IC et de récentes études ont fait émerger l’importance d’un « contrôle qualité » efficace des mitochondries afin de limiter les dommages oxydatifs lors d’un stress cardiaque [1]. Une restauration du système autophagie-lysosome, ou plus particulièrement de la mitophagie apparaît désormais comme un nouvel axe thérapeutique dans l’IC permettant de contrer le stress oxydant, de limiter la déficience énergétique et de prévenir la mort des cardiomyocytes [2]. Cependant, de nombreuses contradictions subsistent dans la compréhension des mécanismes à l’origine de l’altération de l’autophagie au cours d’un stress cardiaque. Dans notre équipe, nous étudions les mécanismes moléculaires associés aux pathologies et au vieillissement cardiaque, afin d’identifier de nouvelles cibles thérapeutiques et d’évaluer de nouvelles interventions pharmacologiques visant à contrer le développement de l’IC (Figure 1). Un de nos objectifs vise à mieux comprendre comment ces événements moléculaires interfèrent avec l’autophagie pour réguler le devenir des cardiomyocytes (mort cellulaire, hypertrophie, sénescence). En améliorant l’autophagie, nous espérons contrer les mécanismes délétères associés au dysfonctionnement cardiaque tels que la mort cellulaire et les dommages mitochondriaux.
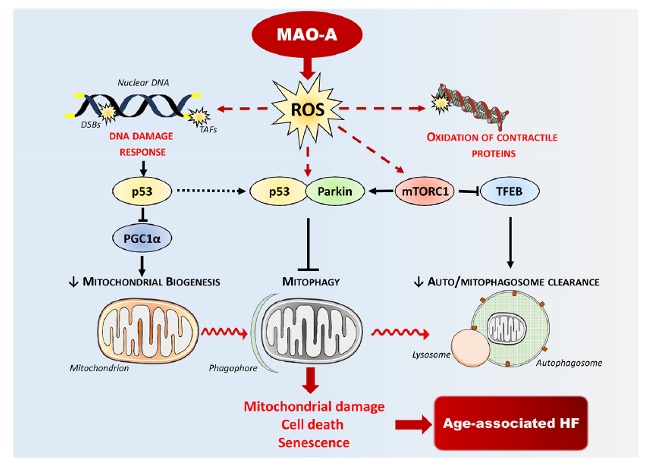
Parmi ces mécanismes nous avons montré que la protéine sensible à l’AMPc Epac1 favorisait l’autophagie aux stades précoces du remodelage cardiaque [3]. Par ailleurs, nous avons caractérisé une nouvelle voie de signalisation couplant la production d’espèces réactives de l’oxygène (EROs) par la monoamine oxydase-A (MAO-A) à la mitophagie (Figure 2). Les MAOs sont des enzymes mitochondriales qui produisent de l’H2O2 lors de la dégradation de leurs substrats, générant un stress oxydant mitochondrial et la mort/sénescence des cardiomyocytes [4, 5]. L’activation chronique de la MAO-A cardiaque au cours du vieillissement inhibe la mitophagie via la protéine parkin et amplifie les dommages mitochondriaux observés dans ce contexte [6, 7].
Une partie de nos travaux vise également à mieux comprendre le lien étroit entre l’accumulation de vésicules autophagiques et la dysfonction mitochondriale dans l’IC. De manière importante, nous avons établi que l’activation de la MAO-A entraine une altération de l’acidification lysosomale associée à un défaut de translocation nucléaire du facteur de transcription (TFEB), un « master » régulateur de l’autophagie et de la biogenèse des lysosomes (Figure 2). In vitro, nous avons montré que les conséquences directes de ces altérations étaient un blocage du flux autophagique, participant à la nécrose des cardiomyocytes [8]. De façon très intéressante, la surexpression de TFEB chez la souris a permis de réduire l’accumulation d’autophagosomes, la fission mitochondriale, la mort des cardiomyocytes ainsi que l’IC chez ces souris. Ainsi, un nouvel axe de recherche s’oriente vers une restauration du « flux autophagique » dans l’IC, via une amélioration de la fusion autophagosome-lysosome ou de la fonction lysosomale. Certains polyphénols semblent intéressants dans ce domaine, via leur rôle activateur de TFEB, et des travaux de l’équipe ont montré que l’Oleuropéine aglycone, un polyphénol extrait de l’huile d’olive extra vierge était un candidat potentiel [9]. En revanche, il existe toujours un manque drastique d’outils lysosomotropes dont l’utilisation pourrait restaurer la fonction des lysosomes dans un contexte pathologique, et c’est dans cet objectif que nos travaux s’orientent à l’heure actuelle.
Figure descriptive

Publications
1. Brown, D.A., et al., Mitochondrial function as a therapeutic target in heart failure. Nature Reviews Cardiology, 2017. 14(4): p. 238-250.
2. Mialet-Perez, J. and C. Vindis, Autophagy in health and disease: focus on the cardiovascular system. Essays in biochemistry, 2017. 61(6): p. 721-732.
3. Laurent, A.-C., et al., Exchange protein directly activated by cAMP 1 promotes autophagy during cardiomyocyte hypertrophy. Cardiovascular research, 2015. 105(1): p. 55-64.
4. Santin, Y., et al., Monoamine oxidases in age-associated diseases: New perspectives for old enzymes. Ageing Res Rev, 2021. 66: p. 101256.
5. Santin, Y., et al., Mitochondrial 4-HNE derived from MAO-A promotes mitoCa(2+) overload in chronic postischemic cardiac remodeling. Cell Death Differ, 2020. 27(6): p. 1907-1923.
6. Anderson, R., et al., Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. Embo j, 2019. 38(5).
7. Manzella, N., et al., Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging cell, 2018. 17(5): p. e12811.
8. Santin, Y., et al., Oxidative Stress by Monoamine Oxidase-A Impairs Transcription Factor EB Activation and Autophagosome Clearance, Leading to Cardiomyocyte Necrosis and Heart Failure. Antioxid Redox Signal, 2016. 25(1): p. 10-27.
9. Miceli, C., et al., Oleuropein Aglycone Protects against MAO-A-Induced Autophagy Impairment and Cardiomyocyte Death through Activation of TFEB. Oxidative medicine and cellular longevity, 2018. 2018: p. 8067592.
Composition de l'équipe
Jeanne Mialet-Perez, CRCN INSERM,
Frank Lezoualc’h, DR INSERMCaroline Conte, MCU
Yannis Sainte-Marie, MCU
Olivier Lairez, PU-PH
Yohan Santin, Post-doc
Karina Formoso, Post-doc
Maximin Détrait, Post-doc
Dorian Bergonnier, Ass. Ing.
Jessica Resta, Doctorante
Loubna Kehal, Doctorante