Physiopathologie des Syndromes Parkinsoniens
Institut des Maladies NeurodégénérativesUniversité de Bordeaux
CNRS UMR 5293 – Bât. Neurocampus
146 Rue Léo Saignat
33076 Bordeaux – France - Bordeaux
Site web - benjamin.dehay@u-bordeaux.fr -
Responsable d'équipe
Benjamin Dehay

Thématiques de recherche
Notre équipe de recherche a pour objectif de comprendre les mécanismes moléculaires et cellulaires responsables des symptômes moteurs et non-moteurs de la maladie de Parkinson et des syndromes parkinsoniens atypiques, ainsi que leur réponse perturbée à la L-Dopa. Notre but est d’identifier de nouvelles cibles thérapeutiques grâce à des études physiopathologiques dans des modèles expérimentaux (allant de la culture cellulaire au primate non-humain) ainsi que chez l’homme.
Notre recherche se développe autour de différents axes :
◦ Mécanisme de mort cellulaire et syndromes parkinsoniens
◦ Modélisation de la dissémination de type prion des synucleinopathies et tauopathies dans le système
nerveux central et périphérique liée aux syndromes parkinsoniens et aux démences.
◦ Recherches sur l’autophagie ciblée comme traitement potentiel de la maladie de Parkinson.
◦ Etude des mécanismes clés contribuant à l’accumulation d’alpha-synucléine pour, à terme, permettre
de contrer la progression de la maladie de Parkinson et de l’atrophie multisystématisée.
◦ Amélioration des approches de transgénèse dans la modélisation des maladies neurodégénératives
◦ Etude du rôle de l’espace extracellulaire dans la communication neuronale
◦ Modulation de la biogenèse lysosomale chez des modèles expérimentaux de la maladie de Parkinson
et de dyskinésie induite par L-dopa
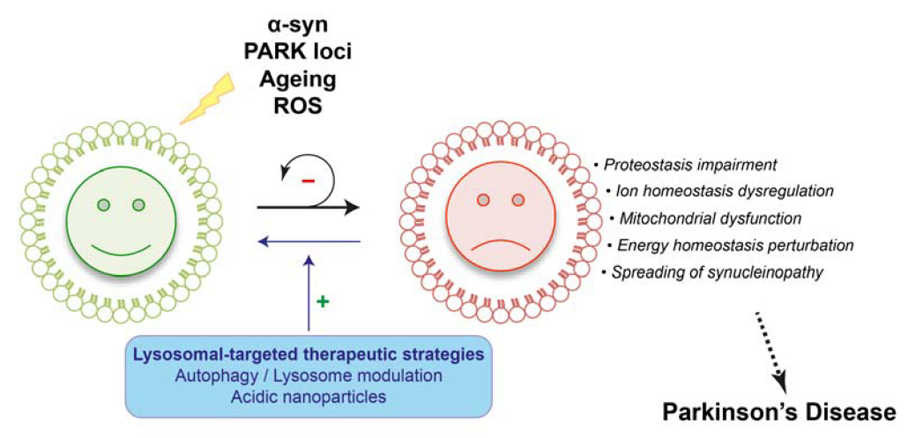
Chez les individus atteints de la maladie de Parkinson, le mécanisme de mort des cellules nerveuses est caractérisé par une accumulation d’-synucléine défectueuse. Notre objectif est de comprendre les processus à l’origine de ce phénomène et de proposer des approches thérapeutiques adéquates. De nombreuses évidences indiquent que l’altération de la fonction autophagie-lysosomale peut contribuer à la pathogenèse de la maladie de Parkinson. En particulier, les déficiences au niveau de la fonction du lysosome pourraient potentiellement rendre compte non seulement du dysfonctionnement et de la mort des cellules neuronales, de la présence des corps de Lewy contenant de l’-synucléine, mais également de la libération de l’α-synucléine par les neurones. Notre objectif principal est de comprendre comment les protéines impliquées dans les maladies neurodégénératives provoquent des altérations des voies protéolytiques contribuant à la pathogenèse et, plus important encore, si l’augmentation fonctionnelle de la fonction et/ou de l’activité lysosomale peut conduire à l’élimination des protéines toxiques et arrêter le processus pathologique. Pour ce deuxième objectif, nous développons de nouvelles stratégies thérapeutiques neuroprotectrices de la maladie visant à rétablir une fonction lysosomale physiologique dans des modèles multidimensionnels et cliniquement pertinents pour les rongeurs et les primates non-humains. En plus des différents projets de recherche, nous commençons l’étude de l’interaction entre l’autophagie et la machinerie ciliaire dans le contexte des maladies neurodégénératives, avec un accent particulier sur la maladie d’Alzheimer.
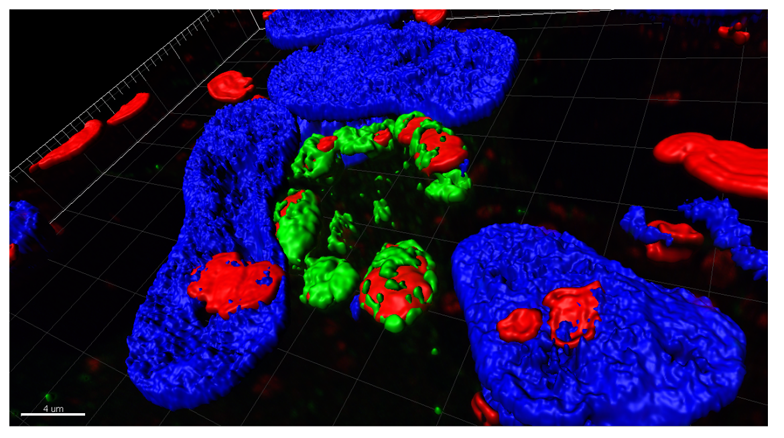
Figure descriptive
Publications
Bourdenx M, Dehay B. Autophagy and brain: specificities and dysfunctions. Med Sci (Paris), volume 33 n°3 (2017).
Bourdenx M, Dehay B. What lysosomes actually tell us about Parkinson’s disease? Ageing Res Rev. 2016 Mar3. pii:S1568-1637(16)30023-X.
Bourdenx M, Daniel J, Genin E, Soria FN, Blanchard-Desce M, Bezard E, Dehay B#. Nanoparticles restore lysosomal acidification defects: Implication for Parkinson’s and other lysosomal-related diseases. Autophagy. 2016 Mar 3;12(3):472-83.
Bourdenx, M., Bezard, E., Dehay, B. Lysosomes and α-synuclein form a dangerous duet leading to neuronal cell death. Front Neuroanat. 2014 Aug 14;8:83.
Bove, J. †, Martinez-Vicente, M. †, Dehay, B. †, Perier, C., Recasens, A., Bombrun, A., Antonsson, B., and Vila, M. Bax channel activity mediates lysosomal disruption linked to Parkinson’s disease. Autophagy. 2014 May;10(5):889-900. († co-first author)
Dehay, B., Martinez-Vicente, M., Caldwell, G., Caldwell, K., Yue, Z., Cookson, M., Klein, C., Vila, M., and Bezard, E. Lysosomal Impairment in Parkinson’s disease. Mov Disorder. 2013 Jun;28(6):725-32.
Dehay, B., Martinez-Vicente, M., Ramirez, A., Perier, C., Klein, C., Vila, M., and Bezard, E. Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy. 2012. Sep;8(9):1389-91.
Dehay, B., Ramirez, A., Martinez-Vicente, M., Perier, C., Canron, MH., Doudnikoff, E., Vital, A., Vila, M., Klein, C., and Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012 Jun 12;109 (24) :9611-6.
Vila, M., Bove, J., Dehay, B., Rodriguez-Muela, C., and Boya, P. Lysosomal membrane permeabilization in Parkinson’s Disease. Autophagy 2011 Jan;7(1):98-100.
Dehay, B., Bove, J., Rodriguez-Muela, C., Perier, C., Recasens, A., Boya, P., and Vila, M. Pathogenic lysosomal depletion in Parkinson’s Disease. J Neurosci. 2010 Sep 15; 30(37): 12535-44.
Composition de l'équipe
AROTCARENA Marie-Laure (doctorante)
BEZARD Erwan (chercheur)
BOUE-GRABOT Eric (chercheur)
BOURDENX Mathieu (post-doctorant)
CANRON Marie-Hélène (assistante ingénieure)
CHANSEL Lucie (doctorante assistante HU)
DEHAY Benjamin (chercheur)
DOVERO Sandra (ingénieure d’étude)
DOUDNIKOFF Marie-Evelyne (assistante ingénieure)
ESTAGER Alain (adjoint technique)
FERNAGUT Pierre-Olivier (chercheur)
GUERIN Paul-Arnaud (doctorant)
JIMENEZ Clément (PU-PH)
LARGITTE Leslie-Ann (ingénieure d’étude)
MARTIN-NEGRIER Marie-Laure (PU-PH)
MARTINEZ Audrey (assistante ingénieure)
MASSE Karine (MCU)
MEISSNER Wassilios (PU-PH)
PAMPLIEGA Olatz (post-doctorante)
SORIA Federico (post-doctorant)
THIOLAT Marie-Laure (technicienne)
TISON François (PU-PH)
VITAL Anne (PU-PH)